- Features: Our gene selection module provides six
supervised algorithms to identify differentially expressed genes
in microarray data based on ordinal or categorical class labels
(five different algorithms and an ensemble approach combining
multiple methods together).
The user can either upload his own microarray data (see uploading your own data) or use one of the
pre-processed example data sets (to obtain more information on
these data sets, please click on the question marks behind the
data set labels).
- Settings: The only parameter that has to be set by
the user is the maximum number of genes to be selected.
Please make sure to inspect the q-values and other significance
scores for the selected genes provided in the output HTML
report - even for a small maximum gene subset size not all
selected genes might be significant hits.
- Output: After
submitting an analysis, an HTML report is generated providing the
results in form of tables and graphs. This includes a ranked list
of differentially expressed genes (each column can be sorted by
clicking on the column title), confidence measures for each
gene (depending on the used algorithm) and boxplots and a heatmap
to visualize the expression values of selected genes across
different samples and sample classes.
If standard gene
identifiers are used in the data (Affymetrix ID, ENTREZ ID,
GENBANK ID, etc.) and the identifiers can be mapped to online
annotation data bases (e.g. ENSEMBLE, DAVID), the selected genes
become hyperlinks which lead the user to the corresponding data
base entries.
If you would like to see an example analysis or obtain more
detailed instructions, please have a look out our video tutorial
section on the main page.
- Uploading your own data: In order to use
ArrayMining.net with your own data there are two possibilities:
Option 1: You can
upload a tab- or
space-delimited text-file containing pre-normalized Microarray
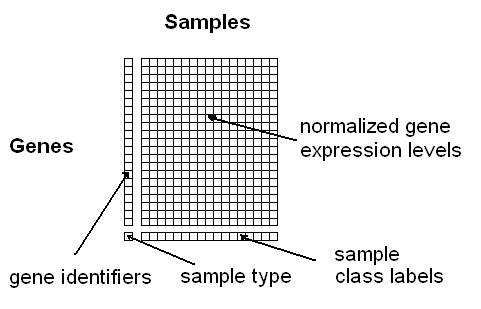
data in the following simple matrix-format (see Fig. 1):
You can download an example data file here (use right-click and "Save as"). The
columns must correspond to the samples and the rows to the genes.
The first column contains the gene identifiers (a unique label
per gene) and the last row the class information for the samples
(multiple samples can have the same class label). The rest of the
matrix should contain normalized expression values obtained using
any of the common Microarray normalization methods (e.g. VSN,
RMA, GCRMA, MAS, dChip, etc.). The gene identifiers can be
any one of the following: Affymetrix ID, ENTREZ ID, GENBANK ID.
You can also use your own identifiers; however, in this case you
won't obtain any links to functional annotation data bases. The
class labels can be any alphanumeric strings or symbols
(e.g. "tumour" and "healthy", or "1","2", "3", or "leukemia1",
"leukemia2", "leukemia3", etc.). Samples belonging to the same
class need to have exactly the same class label. The last row
containing the class labels has to begin with a user-defined
"sample type"-label, e.g. "phenotypes", "tumours" or just
"labels". Optionally, unique IDs per sample can be specified
in the first row (if this line is missing, the samples
will be numbered consecutively).
Option 2: You can upload
a compressed ZIP-archive containing Affymetrix CEL-files
and a txt-file containing tab-delimited numerical sample labels (specifying
replicates by the same number, i.e. "1 1 1 2 2 2" for an experiment
with 6 samples, two classes and three samples for both class 1 and class 2)
Please contact us, should you
experience any kind of problems when uploading or analyzing your
data.
Close
|
 Golub et al. (1999) Leukemia data set
Golub et al. (1999) Leukemia data set 
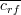 is the average
feature-class correlation and
is the average
feature-class correlation and 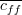 the average feature-feature correlation.
the average feature-feature correlation.